Flow Cytometry Core Facility

The EPFL Flow Cytometry Core Facility (FCCF) provides state-of-the-art instrumentation, technical expertise, theoretical and technical training, and cell sorting service.
FCCF Staff also offers advice and consultancy on experimental design, sample preparation, data acquisition, analysis and interpretation, as well as technical support and troubleshooting.
We are committed to provide comprehensive flow cytometry services in a collaborative environment to help our users and customers to achieve their scientific goals.
Staff of the EPFL FCCF has extensive knowledge and experience in flow cytometry. If you have any questions or if you need help, please contact us or meet us at the facility. Researchers and students are highly encouraged to discuss their ideas and/or needs with us.
Our services and facility are available to internal EPFL customers, but also to members of external Universities and commercial companies.
Find out more
About Us
Who we are and how to find the facility.
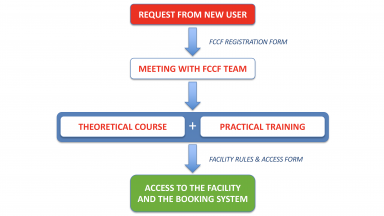
Getting Started
Your starting point if you want to use our equipment.

Instruments
Equipment available at the facility.
Contact
We are located in the AI Building, adjacent to the SV Building, in the western part of the EPFL campus.
EPFL SV PTCF
AI 0247
AI Building
Station 15
CH-1015 Lausanne
For any flow cytometry request, please contact us via the following email address:
For any mass cytometry request, please use the following email address: